Tutorial: Basic Definitions and Settings¶
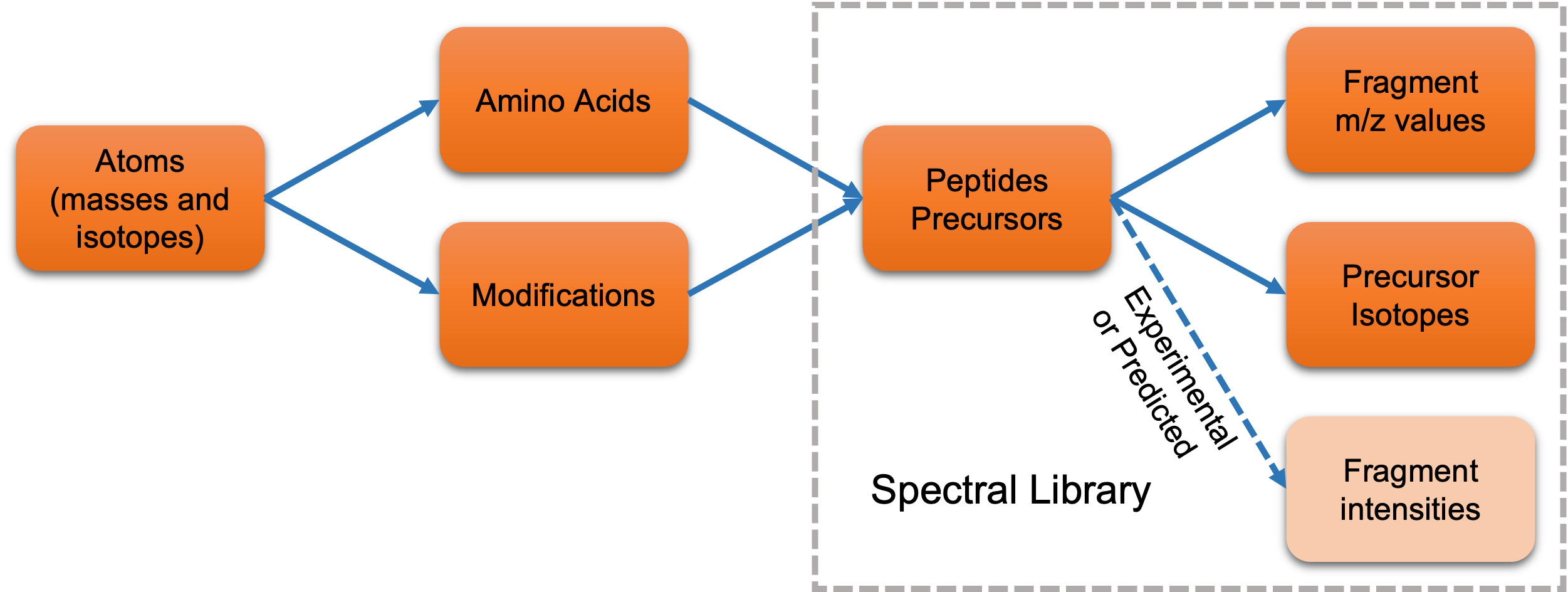
Measuring m/z values is the elemental function of MS technologies, therefore the calculation of mass values for a peptide and its fragments becomes the most essential part in MS-based computational tools. AlphaBase calculates all mass values from atoms. And the masses of amino acids and modifications are calculated from their atom compositions, repectively. Eventually, the masses of peptides or precursors as well as their fragments can be calculated by the amino acid sequences with or without modifications (See figure below).
Calculating masses from atoms makes it much easier to switch between unlabeled and heavy-labeled peptides, as we did in Stellar MS for 15N-labeled peptides as the reference for targeted proteomics (https://www.biorxiv.org/content/10.1101/2024.06.02.597029v2.full).
The other advantage of starting from atoms is that AlphaBase can calculate isotope distributions of peptides based on a pre-defined isotope distribution list of atoms (e.g., NIST atom table in https://physics.nist.gov/cgi-bin/Compositions/stand_alone.pl). The isotope information has been applied in our alphaDIA search engine to boost the identification of DIA-MS data (https://www.biorxiv.org/content/10.1101/2024.05.28.596182v1).
Atoms/Elements¶
The masses of all amino acids and modifications are calculated from their atom compositions.
The atom information are defined in https://github.com/MannLabs/alphabase/blob/main/alphabase/constants/const_files/nist_element.yaml which is parsed from NIST, see https://github.com/MannLabs/alphabase/blob/main/scripts/nist_chem_to_yaml.ipynb.
After adding some heavy isotopes, including 13C, 15N, 2H, and 18O, we obtain 109 kinds of atoms:
[1]:
import pandas as pd
from alphabase.constants.atom import CHEM_INFO_DICT
pd.DataFrame().from_dict(CHEM_INFO_DICT, orient='index')
[1]:
| abundance | mass | |
|---|---|---|
| 13C | [0.01, 0.99] | [12.0, 13.00335483507] |
| 14N | [0.996337, 0.003663] | [14.00307400443, 15.00010889888] |
| 15N | [0.01, 0.99] | [14.00307400443, 15.00010889888] |
| 18O | [0.005, 0.005, 0.99] | [15.99491461957, 16.9991317565, 17.99915961286] |
| 2H | [0.01, 0.99] | [1.00782503223, 2.01410177812] |
| ... | ... | ... |
| Xe | [0.000952, 0.00089, 0.019102, 0.264006, 0.0407... | [123.905892, 125.9042983, 127.903531, 128.9047... |
| Y | [1.0] | [88.9058403] |
| Yb | [0.00123, 0.02982, 0.1409, 0.2168, 0.16103, 0.... | [167.9338896, 169.9347664, 170.9363302, 171.93... |
| Zn | [0.4917, 0.2773, 0.0404, 0.1845, 0.0061] | [63.92914201, 65.92603381, 66.92712775, 67.924... |
| Zr | [0.5145, 0.1122, 0.1715, 0.1738, 0.028] | [89.9046977, 90.9056396, 91.9050347, 93.906310... |
109 rows × 2 columns
And their mono-isotopic mass are in CHEM_MONO_MASS (dict):
[2]:
from alphabase.constants.atom import CHEM_MONO_MASS
pd.DataFrame().from_dict(CHEM_MONO_MASS, orient='index')
[2]:
| 0 | |
|---|---|
| 13C | 13.003355 |
| 14N | 14.003074 |
| 15N | 15.000109 |
| 18O | 17.999160 |
| 2H | 2.014102 |
| ... | ... |
| Xe | 131.904155 |
| Y | 88.905840 |
| Yb | 173.938866 |
| Zn | 63.929142 |
| Zr | 89.904698 |
109 rows × 1 columns
These atom masses are used to calculate the masses of amino acids, modifications, and then subsequent masses of peptides and fragments.
Commonly used molecular masses¶
[3]:
from alphabase.constants.atom import (
MASS_PROTON, MASS_ISOTOPE, MASS_NH3, MASS_H2O
)
MASS_PROTON, MASS_ISOTOPE, MASS_NH3, MASS_H2O
[3]:
(1.007276467, 1.0033, 17.02654910112, 18.01056468403)
Amino Acids¶
[4]:
from alphabase.constants.aa import AA_DF
AA_DF.loc[ord('A'):ord('Z')]
[4]:
| aa | formula | smiles | mass | |
|---|---|---|---|---|
| 65 | A | C(3)H(5)N(1)O(1)S(0) | N([Fl])([Fl])[C@@]([H])(C)C(=O)[Ts] | 7.103711e+01 |
| 66 | B | C(1000000) | NaN | 1.200000e+07 |
| 67 | C | C(3)H(5)N(1)O(1)S(1) | N([Fl])([Fl])[C@@]([H])(CS)C(=O)[Ts] | 1.030092e+02 |
| 68 | D | C(4)H(5)N(1)O(3)S(0) | N([Fl])([Fl])[C@@]([H])(CC(=O)O)C(=O)[Ts] | 1.150269e+02 |
| 69 | E | C(5)H(7)N(1)O(3)S(0) | N([Fl])([Fl])[C@@]([H])(CCC(=O)O)C(=O)[Ts] | 1.290426e+02 |
| 70 | F | C(9)H(9)N(1)O(1)S(0) | N([Fl])([Fl])[C@@]([H])(Cc1ccccc1)C(=O)[Ts] | 1.470684e+02 |
| 71 | G | C(2)H(3)N(1)O(1)S(0) | N([Fl])([Fl])CC(=O)[Ts] | 5.702146e+01 |
| 72 | H | C(6)H(7)N(3)O(1)S(0) | N([Fl])([Fl])[C@@]([H])(CC1=CN=C-N1)C(=O)[Ts] | 1.370589e+02 |
| 73 | I | C(6)H(11)N(1)O(1)S(0) | N([Fl])([Fl])[C@@]([H])([C@]([H])(CC)C)C(=O)[Ts] | 1.130841e+02 |
| 74 | J | C(6)H(11)N(1)O(1)S(0) | NaN | 1.130841e+02 |
| 75 | K | C(6)H(12)N(2)O(1)S(0) | N([Fl])([Fl])[C@@]([H])(CCCCN)C(=O)[Ts] | 1.280950e+02 |
| 76 | L | C(6)H(11)N(1)O(1)S(0) | N([Fl])([Fl])[C@@]([H])(CC(C)C)C(=O)[Ts] | 1.130841e+02 |
| 77 | M | C(5)H(9)N(1)O(1)S(1) | N([Fl])([Fl])[C@@]([H])(CCSC)C(=O)[Ts] | 1.310405e+02 |
| 78 | N | C(4)H(6)N(2)O(2)S(0) | N([Fl])([Fl])[C@@]([H])(CC(=O)N)C(=O)[Ts] | 1.140429e+02 |
| 79 | O | C(12)H(19)N(3)O(2) | C[C@@H]1CC=N[C@H]1C(=O)NCCCC[C@@H](C(=O)[Ts])N... | 2.371477e+02 |
| 80 | P | C(5)H(7)N(1)O(1)S(0) | N1([Fl])[C@@]([H])(CCC1)C(=O)[Ts] | 9.705276e+01 |
| 81 | Q | C(5)H(8)N(2)O(2)S(0) | N([Fl])([Fl])[C@@]([H])(CCC(=O)N)C(=O)[Ts] | 1.280586e+02 |
| 82 | R | C(6)H(12)N(4)O(1)S(0) | N([Fl])([Fl])[C@@]([H])(CCCNC(=N)N)C(=O)[Ts] | 1.561011e+02 |
| 83 | S | C(3)H(5)N(1)O(2)S(0) | N([Fl])([Fl])[C@@]([H])(CO)C(=O)[Ts] | 8.703203e+01 |
| 84 | T | C(4)H(7)N(1)O(2)S(0) | N([Fl])([Fl])[C@@]([H])([C@]([H])(O)C)C(=O)[Ts] | 1.010477e+02 |
| 85 | U | C(3)H(5)N(1)O(1)Se(1) | N([Fl])([Fl])[C@@]([H])(C[Se][H])C(=O)[Ts] | 1.509536e+02 |
| 86 | V | C(5)H(9)N(1)O(1)S(0) | N([Fl])([Fl])[C@@]([H])(C(C)C)C(=O)[Ts] | 9.906841e+01 |
| 87 | W | C(11)H(10)N(2)O(1)S(0) | N([Fl])([Fl])[C@@]([H])(CC(=CN2)C1=C2C=CC=C1)C... | 1.860793e+02 |
| 88 | X | C(1000000) | NaN | 1.200000e+07 |
| 89 | Y | C(9)H(9)N(1)O(2)S(0) | N([Fl])([Fl])[C@@]([H])(Cc1ccc(O)cc1)C(=O)[Ts] | 1.630633e+02 |
| 90 | Z | C(1000000) | NaN | 1.200000e+07 |
In AA_DF, amino acids are encoded by ASCII (128 characters), thus 65==ord(‘A’), …, 90==ord(‘Z’). Unicode strings can be quickly converted to ASCII int32 values using np.array.view():
[5]:
import numpy as np
np.array(['ABCXYZ']).view(np.int32)
[5]:
array([65, 66, 67, 88, 89, 90], dtype=int32)
But users does not need to know this, as we provided easy to use functionalities to get residue masses from sequences.
Calculate AA masses in batch¶
[6]:
from alphabase.constants.aa import calc_AA_masses_for_same_len_seqs
calc_AA_masses_for_same_len_seqs(
[
'MACDEFG', 'MAKDEFG', 'MAKDEFR'
]
)
[6]:
array([[131.04048509, 71.03711379, 103.00918496, 115.02694302,
129.04259309, 147.06841391, 57.02146372],
[131.04048509, 71.03711379, 128.09496302, 115.02694302,
129.04259309, 147.06841391, 57.02146372],
[131.04048509, 71.03711379, 128.09496302, 115.02694302,
129.04259309, 147.06841391, 156.10111102]])
[7]:
from alphabase.constants.aa import calc_AA_masses_for_var_len_seqs
calc_AA_masses_for_var_len_seqs(
[
'M', 'MAK', 'MAKDEFR'
])
[7]:
array([[1.31040485e+02, 1.00000000e+08, 1.00000000e+08, 1.00000000e+08,
1.00000000e+08, 1.00000000e+08, 1.00000000e+08],
[1.31040485e+02, 7.10371138e+01, 1.28094963e+02, 1.00000000e+08,
1.00000000e+08, 1.00000000e+08, 1.00000000e+08],
[1.31040485e+02, 7.10371138e+01, 1.28094963e+02, 1.15026943e+02,
1.29042593e+02, 1.47068414e+02, 1.56101111e+02]])
Modifications¶
In AlphaBase, we used mod_name@aa to represent a modification, the mod_name is from UniMod. We also used mod_name@Protein_N-term, mod_name@Any_N-term and mod_name@Any_C-term for terminal modifications, which follow the UniMod terminal name schema.
The default modification TSV is stored in https://github.com/MannLabs/alphabase/blob/main/alphabase/constants/const_files/modification.tsv, which is loaded upon startup of AlphaBase. Users can add more modifications into the tsv file (only mod_name and composition columns are required), e.g. by using the https://github.com/MannLabs/alphabase/blob/main/scripts/unimod_to_tsv.ipynb notebook.
[8]:
from alphabase.constants.modification import MOD_DF
MOD_DF
[8]:
| mod_name | unimod_mass | unimod_avge_mass | composition | unimod_modloss | modloss_composition | classification | unimod_id | smiles | modloss_importance | mass | modloss_original | modloss | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mod_name | |||||||||||||
| Acetyl@T | Acetyl@T | 42.010565 | 42.0367 | H(2)C(2)O(1) | 0.0 | Post-translational | 1 | 0.0 | 42.010565 | 0.0 | 0.0 | ||
| Acetyl@Protein_N-term | Acetyl@Protein_N-term | 42.010565 | 42.0367 | H(2)C(2)O(1) | 0.0 | Post-translational | 1 | CC(=O)[Ts] | 0.0 | 42.010565 | 0.0 | 0.0 | |
| Acetyl@S | Acetyl@S | 42.010565 | 42.0367 | H(2)C(2)O(1) | 0.0 | Post-translational | 1 | 0.0 | 42.010565 | 0.0 | 0.0 | ||
| Acetyl@C | Acetyl@C | 42.010565 | 42.0367 | H(2)C(2)O(1) | 0.0 | Post-translational | 1 | 0.0 | 42.010565 | 0.0 | 0.0 | ||
| Acetyl@Any_N-term | Acetyl@Any_N-term | 42.010565 | 42.0367 | H(2)C(2)O(1) | 0.0 | Multiple | 1 | CC(=O)[Ts] | 0.0 | 42.010565 | 0.0 | 0.0 | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Lactyl@Any_N-term | Lactyl@Any_N-term | 72.021129 | 72.0627 | H(4)C(3)O(2) | 0.0 | Post-translational | 0 | C[C@@H](O)C(=O)[Ts] | 0.0 | 72.021129 | 0.0 | 0.0 | |
| Lactyl@Protein_N-term | Lactyl@Protein_N-term | 72.021129 | 72.0627 | H(4)C(3)O(2) | 0.0 | Post-translational | 0 | C[C@@H](O)C(=O)[Ts] | 0.0 | 72.021129 | 0.0 | 0.0 | |
| YnLactyl@K | YnLactyl@K | 239.126991 | 239.2941 | H(17)C(11)N(3)O(3) | 0.0 | Post-translational | 0 | OCCCCCCN1C=C(C[C@@H](O)C(=O)NCCCC[C@H](N([Fl])... | 0.0 | 239.126991 | 0.0 | 0.0 | |
| YnLactyl@Any_N-term | YnLactyl@Any_N-term | 239.126991 | 239.2941 | H(17)C(11)N(3)O(3) | 0.0 | Post-translational | 0 | OCCCCCCN1C=C(C[C@@H](O)C(=O)[Ts])N=N1 | 0.0 | 239.126991 | 0.0 | 0.0 | |
| YnLactyl@Protein_N-term | YnLactyl@Protein_N-term | 239.126991 | 239.2941 | H(17)C(11)N(3)O(3) | 0.0 | Post-translational | 0 | OCCCCCCN1C=C(C[C@@H](O)C(=O)[Ts])N=N1 | 0.0 | 239.126991 | 0.0 | 0.0 |
2852 rows × 13 columns
Modification sites¶
In alphabase, we use 0 and -1 to represent modification site of N-term and C-term, respectively. For other modification sites, we use 1 to n.
[9]:
from alphabase.constants.modification import calc_modification_mass
# example: add two modifications and print the array of mass modifications
sequence = 'MACDEFG'
mod_names = ['Acetyl@Any_N-term', 'Carbamidomethyl@C']
mod_sites = [0, 3] # 0 for N-term, 3 for the third amino acid
calc_modification_mass(
nAA=len(sequence),
mod_names=mod_names,
mod_sites=mod_sites
)
[9]:
array([42.01056468, 0. , 57.02146372, 0. , 0. ,
0. , 0. ])
[10]:
# example: add two modifications and print the array of mass modifications
sequence = 'MAKDEFG'
mod_names = ['Acetyl@Any_N-term', 'Oxidation@M']
mod_sites = [0, 1] # 0 for N-term, 1 for the first amino acid
calc_modification_mass(
nAA=len(sequence),
mod_names=mod_names,
mod_sites=mod_sites
)
[10]:
array([58.0054793, 0. , 0. , 0. , 0. ,
0. , 0. ])
Multiple modifications at a single site is supported, for example, in the following example, K3 contains both GG@K and Dimethyl@K:
[11]:
sequence = 'MAKDEFR'
mod_names = ['GG@K', 'Dimethyl@K']
mod_sites = [3, 3]
calc_modification_mass(
nAA=len(sequence),
mod_names=mod_names,
mod_sites=mod_sites
)
[11]:
array([ 0. , 0. , 142.07422757, 0. ,
0. , 0. , 0. ])
Caculate modification masses in batch¶
[12]:
from alphabase.constants.modification import calc_mod_masses_for_same_len_seqs
calc_mod_masses_for_same_len_seqs(
nAA=7,
mod_names_list=[
['Acetyl@Any_N-term', 'Carbamidomethyl@C'],
['Acetyl@Any_N-term', 'Oxidation@M'],
['GG@K', 'Dimethyl@K'],
],
mod_sites_list=[
[0, 3],
[0, 1],
[3, 3],
]
)
[12]:
array([[ 42.01056468, 0. , 57.02146372, 0. ,
0. , 0. , 0. ],
[ 58.0054793 , 0. , 0. , 0. ,
0. , 0. , 0. ],
[ 0. , 0. , 142.07422757, 0. ,
0. , 0. , 0. ]])
Mass calculation functionalities¶
Calculate AA and modification masses in batch¶
[13]:
from alphabase.constants.aa import calc_AA_masses_for_same_len_seqs
from alphabase.constants.modification import calc_mod_masses_for_same_len_seqs
mod_masses = calc_mod_masses_for_same_len_seqs(
nAA=7,
mod_names_list=[
['Acetyl@Any_N-term', 'Carbamidomethyl@C'],
['Acetyl@Any_N-term', 'Oxidation@M'],
['GG@K', 'Dimethyl@K'],
],
mod_sites_list=[
[0, 3],
[0, 1],
[3, 3],
]
)
aa_masses = calc_AA_masses_for_same_len_seqs(
[
'MACDEFG', 'MAKDEFG', 'MAKDEFR'
]
)
mod_masses+aa_masses
[13]:
array([[173.05104977, 71.03711379, 160.03064868, 115.02694302,
129.04259309, 147.06841391, 57.02146372],
[189.04596439, 71.03711379, 128.09496302, 115.02694302,
129.04259309, 147.06841391, 57.02146372],
[131.04048509, 71.03711379, 270.16919059, 115.02694302,
129.04259309, 147.06841391, 156.10111102]])
np.cumsum to get b-ion neutral masses¶
[14]:
import numpy as np
np.cumsum(aa_masses+mod_masses, axis=1)
[14]:
array([[ 173.05104977, 244.08816356, 404.11881224, 519.14575526,
648.18834835, 795.25676227, 852.27822599],
[ 189.04596439, 260.08307818, 388.17804119, 503.20498422,
632.24757731, 779.31599122, 836.33745494],
[ 131.04048509, 202.07759887, 472.24678946, 587.27373248,
716.31632557, 863.38473949, 1019.48585051]])
Mass functionalities in ‘mass_calc’¶
The functionalities for peptide and fragment neutral masses have been implement in alphabase.peptide.mass_calc:
[15]:
from alphabase.peptide.mass_calc import calc_peptide_masses_for_same_len_seqs
peptide_masses = calc_peptide_masses_for_same_len_seqs(
['MACDEFG', 'MAKDEFG', 'MAKDEFR'],
mod_list=[
'Acetyl@Any_N-term;Carbamidomethyl@C',
'Acetyl@Any_N-term;Oxidation@M',
'GG@K;Dimethyl@K',
],
)
peptide_masses
[15]:
array([ 870.28879067, 854.34801962, 1037.49641519])
[16]:
from alphabase.peptide.mass_calc import calc_b_y_and_peptide_masses_for_same_len_seqs
b_masses, y_masses, peptide_masses = calc_b_y_and_peptide_masses_for_same_len_seqs(
['MACDEFG', 'MAKDEFG', 'MAKDEFR'],
mod_list=[
['Acetyl@Any_N-term', 'Carbamidomethyl@C'],
['Acetyl@Any_N-term', 'Oxidation@M'],
['GG@K', 'Dimethyl@K'],
],
site_list=[
[0, 3],
[0, 1],
[3, 3],
],
)
peptide_masses
[16]:
array([ 870.28879067, 854.34801962, 1037.49641519])
[17]:
b_masses
[17]:
array([[173.05104977, 244.08816356, 404.11881224, 519.14575526,
648.18834835, 795.25676227],
[189.04596439, 260.08307818, 388.17804119, 503.20498422,
632.24757731, 779.31599122],
[131.04048509, 202.07759887, 472.24678946, 587.27373248,
716.31632557, 863.38473949]])
[18]:
y_masses
[18]:
array([[697.2377409 , 626.20062711, 466.16997843, 351.14303541,
222.10044232, 75.0320284 ],
[665.30205523, 594.26494145, 466.16997843, 351.14303541,
222.10044232, 75.0320284 ],
[906.45593011, 835.41881632, 565.24962574, 450.22268271,
321.18008962, 174.11167571]])
Isotope distribution¶
alphabase.constants.isotope.IsotopeDistribution will calculate the isotope distribution and the mono-isotopic idx in the distribution for a given atom composition.
For an atom, the mono-isotopic idx (mono_idx) points to the highest abundance isotope, so the value is round(mass of highest isotope - mass of first isotope).
[19]:
import pandas as pd
from alphabase.constants.atom import CHEM_INFO_DICT
atom_df = pd.DataFrame().from_dict(CHEM_INFO_DICT, orient='index')
def get_mono(masses_abundances):
masses, abundances = masses_abundances
return round(masses[np.argmax(abundances)]-masses[0])
atom_df['mono_idx'] = atom_df[['mass','abundance']].apply(
get_mono, axis=1
)
atom_df
[19]:
| abundance | mass | mono_idx | |
|---|---|---|---|
| 13C | [0.01, 0.99] | [12.0, 13.00335483507] | 1 |
| 14N | [0.996337, 0.003663] | [14.00307400443, 15.00010889888] | 0 |
| 15N | [0.01, 0.99] | [14.00307400443, 15.00010889888] | 1 |
| 18O | [0.005, 0.005, 0.99] | [15.99491461957, 16.9991317565, 17.99915961286] | 2 |
| 2H | [0.01, 0.99] | [1.00782503223, 2.01410177812] | 1 |
| ... | ... | ... | ... |
| Xe | [0.000952, 0.00089, 0.019102, 0.264006, 0.0407... | [123.905892, 125.9042983, 127.903531, 128.9047... | 8 |
| Y | [1.0] | [88.9058403] | 0 |
| Yb | [0.00123, 0.02982, 0.1409, 0.2168, 0.16103, 0.... | [167.9338896, 169.9347664, 170.9363302, 171.93... | 6 |
| Zn | [0.4917, 0.2773, 0.0404, 0.1845, 0.0061] | [63.92914201, 65.92603381, 66.92712775, 67.924... | 0 |
| Zr | [0.5145, 0.1122, 0.1715, 0.1738, 0.028] | [89.9046977, 90.9056396, 91.9050347, 93.906310... | 0 |
109 rows × 3 columns
mono_idx of an atom composition refers to the sum of the mono_idx of all atoms. In AlphaBase, alphabase.constants.isotope.IsotopeDistribution calculate both isotope abundance and mono_idx.
For example, Fe’s mono_idx is 2 (mass from 53.94 to 55.93),
[20]:
atom_df.loc['Fe']
[20]:
abundance [0.05845, 0.91754, 0.02119, 0.00282]
mass [53.93960899, 55.93493633, 56.93539284, 57.933...
mono_idx 2
Name: Fe, dtype: object
So C(1)Fe(1)’s mono_idx is also 2:
[21]:
from alphabase.constants.isotope import IsotopeDistribution, parse_formula
iso = IsotopeDistribution()
iso.calc_formula_distribution(
[('C',1),('Fe',1)]
)
[21]:
(array([5.78245850e-02, 6.25415000e-04, 9.07722322e-01, 3.07809450e-02,
3.01655900e-03, 3.01740000e-05, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00]),
2)
But 13C(1)Fe(1)’s mono_idx should be 3:
[22]:
iso.calc_formula_distribution(
[('13C',1),('Fe',1)]
)
[22]:
(array([5.845000e-04, 5.786550e-02, 9.175400e-03, 9.085765e-01,
2.100630e-02, 2.791800e-03, 0.000000e+00, 0.000000e+00,
0.000000e+00, 0.000000e+00]),
3)
The mono_idx of unlabeled atom compositions is always 0, no matter how big the compositions are. This means mono isotope is not necessary to be the highest isotope peak, especially when the composition get larger. Here are three examples from small composition to large ones, we can see that the highest peaks move from 0 to 2.
[23]:
from alphabase.constants.isotope import IsotopeDistribution, parse_formula
iso = IsotopeDistribution()
formula = 'C(50)H(50)O(20)Na(1)'
formula = parse_formula(formula)
dist, mono = iso.calc_formula_distribution(formula)
f"mono={mono}, highest={dist.argmax()}", dist
[23]:
('mono=0, highest=0',
array([5.53058051e-01, 3.06480210e-01, 1.06031073e-01, 2.73885413e-02,
5.79597328e-03, 1.05055134e-03, 1.67897345e-04, 2.41173838e-05,
3.15729577e-06, 3.80635657e-07]))
[24]:
formula = 'C(100)H(100)O(20)Na(1)'
formula = parse_formula(formula)
dist, mono = iso.calc_formula_distribution(formula)
f"mono={mono}, highest={dist.argmax()}", dist
[24]:
('mono=0, highest=1',
array([3.21124792e-01, 3.53459703e-01, 2.05844502e-01, 8.38383715e-02,
2.66913129e-02, 7.04911613e-03, 1.60206285e-03, 3.21190201e-04,
5.78218885e-05, 9.47198919e-06]))
[25]:
formula = 'C(200)H(200)O(40)Na(1)'
formula = parse_formula(formula)
dist, mono = iso.calc_formula_distribution(formula)
f"mono={mono}, highest={dist.argmax()}", dist
[25]:
('mono=0, highest=2',
array([0.10312113, 0.22700935, 0.25713731, 0.19936063, 0.11878142,
0.05791123, 0.02402947, 0.00871637, 0.00281814, 0.00082412]))
[ ]: